Doutorado: Epidemiologia e História da Ataxia Cerebelar Dominante e Doença de Machado-Joseph no Brasil
 |
Desenho do Estudo. Neste trabalho estudamos, pela primeira vez no Brasil, a epidemiologia e a genética molecular relativas a um grupo de doenças raras conhecidas como ataxia cerebelar autossômica dominante (ACAD). Ele é resultado de 5,8 anos de estudos iniciados por Sousa PS., 1992, com a caracterização clínica de 11 famílias afetadas pela doença. Esses estudos, posteriormente ampliados pelo autor da presente tese, foram inicialmente desenvolvidos na USP de Ribeirão Preto, e mais tarde passaram a abarcar a área de genética molecular da Universidade de Sydney, Austrália. No período de 1990 a 1997 estudamos a distribuição geográfica dessas famílias, às quais foram adicionadas outras, incluindo trabalho de campo em um raio aproximado de 400 km de Ribeirão Preto. Nesse estudo de campo fizemos análise genealógica e clínica das famílias, assim como realizamos coleta de sangue para os estudos de genética molecular desenvolvidos na Universidade mencionada, na Salpetriere em Paris (Stevanim et al., 1994, 1995a, 1995b), e um estudo colaborativo internacional (Igarashi S., et al 1996). O link para o download da tese está nas referências, ao final deste capítulo.
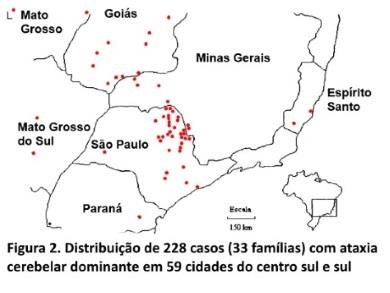
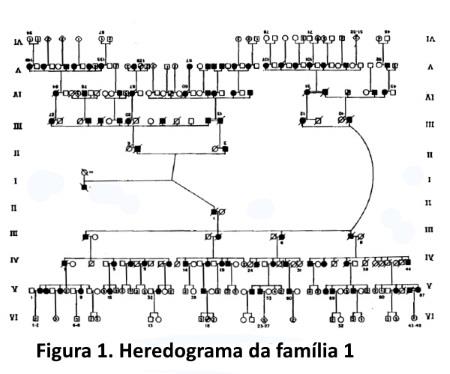
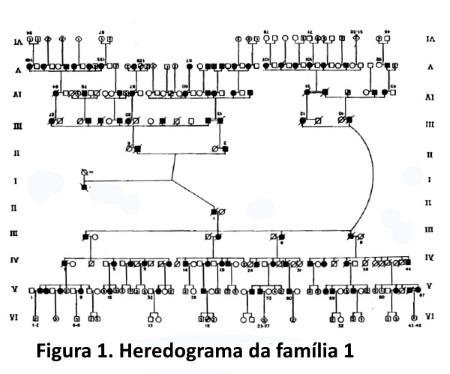
Caracterização das famílias. Num intervalo de 5,8 anos, iniciado em agosto de 1990, foram estudadas 33 famílias com ACAD, compreendendo um total de 2.252 indivíduos (entre vivos e mortos), dos quais 426 são afetados; a maior família estudada tem um total de 46 afetados examinados (figura 1).
O estudo genealógico retrocedido no máximo até 1820 caracterizou um grupo extracontinental com 6 famílias, de procedência Italiana, portuguesa, polonesa e japonesa, e um grupo continental com 27 famílias (compreendendo 93% dos afetados). Nessas famílias a investigação do primeiro ancestral (fundador) indica estarem eles vivendo no Brasil desde 1723, o que não demonstra, assim, uma migração de outro país, embora isso fosse provável. A partir da identificação, tais famílias foram denominadas continentais.
De 186 pacientes examinados, com idade média de 50,7 anos (15-82), em 171 a doença iniciou-se em média aos 41,3 anos (2-76). O fenótipo dessas famílias era formado por ataxia cerebelar (100%), sinais piramidais (94%), oftalmoplegia (88%), hipopalestesia distal (82%) e amiotrofia distal (58%). Achados menos frequentes foram: olhos proeminentes, sinais extrapiramidais e mioquimia facial, ocorrendo cada um em mais de 42% das famílias. Nenhuma delas apresentou demência, distrofia maculopigmentar, mioclonias, ataxia pura nem paroxística.
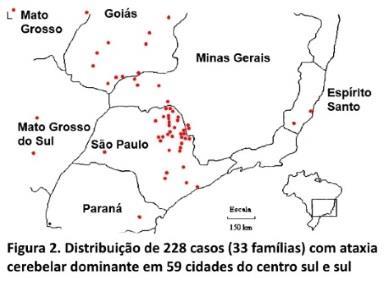 |
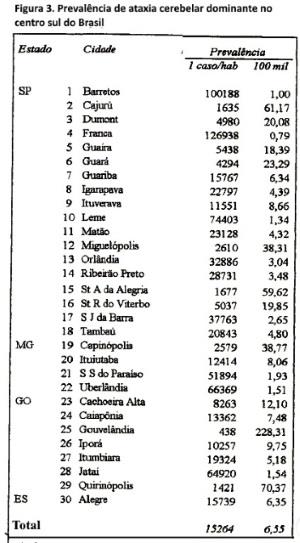
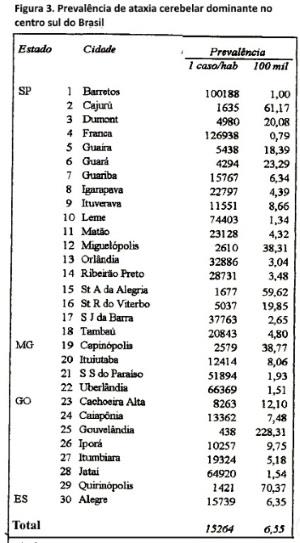
Distribuição geográfica e prevalência. Os 228 pacientes vivos (em 1/07/95), distribuíam-se em 59 cidades do centro-sul do Brasil (figura 2), predominando nos estados de São Paulo (sul do Rio Grande), Goiás (sul) e Minas Gerais (oeste e sul). Em 30 dessas cidades a prevalência (figura 3) média mínima das ACADs foi de 6,5 casos/100.000 habitantes (136 casos/2.075.948 habitantes); prevalência entre 6-228 casos/100.000 habitantes, com existência em 17 cidades.
A presença de um efeito fundador nas famílias continentais é muito provável, pelos seguintes argumentos: 1) a elevada prevalência tanto de casos quanto de números de famílias vivendo em pequenas comunidades; 2) a origem de grupos de famílias a partir de uma única pequena vila; e 3) a origem de várias famílias a partir de vilas muito próximas, anteriormente ao ano de 1830. Pelo exposto, torna-se possível dizer que é provável que as 27 famílias continentais possam ser reduzidas a um número máximo de 14 fundadores (14 famílias).
 |
Dispersão geográfica. A análise de 175 anos de migrações evidenciou que a maioria das famílias continentais assentaram-se nos estados de São Paulo, Minas Gerais e Goiás, a partir do final do século XVIII, provenientes da região da Comarca do Rio das Mortes e da região mineradora do Estado de Minas Gerais. A grande área mineradora (donde provém a família GC 1) teve seu povoamento iniciado em 1674, considerado no século XVII um tipo de El Dorado, atraindo uma população variada proveniente da costa brasileira, assim como portugueses, incluindo açorianos.
A máxima produção de ouro foi alcançada na metade século XVIII, quando a atividade começou a entrar em decadência e as famílias iniciaram um processo de migração para oeste, processo denominado pelos historiadores como “marcha para oeste”. Os primeiros estabelecimentos dessas famílias migradas de Minas Gerais foram as grandes fazendas de criação de gado, notadamente no noroeste de São Paulo, posteriormente alcançando o centro-sul do Brasil. No início das migrações, o acesso migratório a Goiás e a Mato Grosso se deu através do caminho de Anhanguera.
Caracterização molecular. Na família GC1 a análise molecular excluiu expansão no cromossoma 6p, assim como ligação com o cromossoma 6p (D6S89) e 12p (PLA2, IGF1 e D12S84). O gene mutante dessa família foi localizado em 14q, no intervalo entre D14S68 (lod score máximo de 3,53 em O =0) e D14S81 (lod score máximo de 2,23 em 8= 0,05). Estudos de expansão em 14q confirmam a doença de Machado e Joseph em 4 famílias (10,8% dos casos vivos) da presente série (Stevanin G et al., 1995).
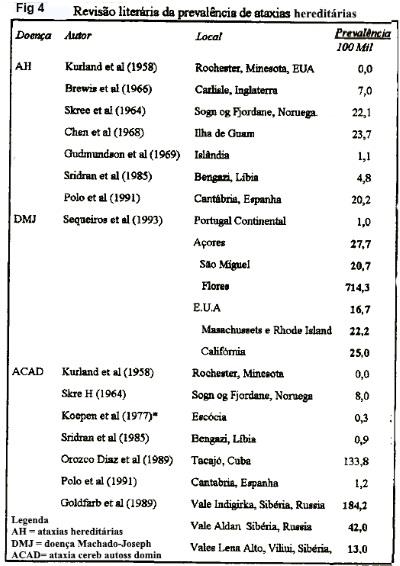 |
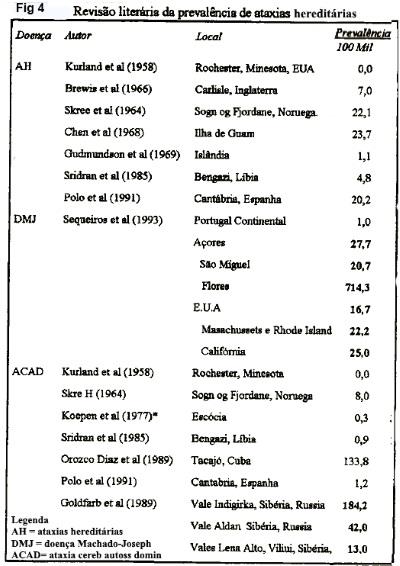
Implicações do presente estudo. Comparada a taxa de prevalência máxima da literatura (figura 4), as ACADS no Brasil (228 casos /100 mil) encontram-se entre as maiores da literatura, incluindo as da Ilha de Guan, com 23 casos/100mil (Chen et al., 1968); do Vale da Indigirka, na Sibéria, com 188 casos/100 mil (Goldfarb et al., 1989); ficando apenas abaixo daquela DMJ na Ilha de Flores, com 714 casos/100 mil (Sequeiros et al., 1993). Restringindo as cifras ao domínio das doenças hereditárias e neurodegenerativas, podemos concluir que as ACADs representam urna condição importante na população brasileira, notadamente nas regiões do centro-sul.
O fenótipo clínico das famílias descritas, a grande ascendência portuguesa-açoriana na população brasileira, a elevada prevalência encontrada e a confirmação molecular da DMJ em algumas famílias sugerem que a DMJ deve ser a ACAD predominante em nossa população. Entretanto, o diagnóstico da DMJ deve ser confirmado por testes de expansão em nível de 14q, em primeiro lugar devido à inespecificidade do fenótipo clínico, e em segundo lugar pela grande diversificação da população brasileira. Embora basicamente formada pelo elemento português, a população brasileira, após 1500, vem sendo diversificada por diferentes elementos étnicos, como Indígenas, negros, Italianos, espanhóis, judeus, franceses, mouros (provavelmente), poloneses e alemães, em alguns desses grupos já tendo sido descritos diferentes loci de ACAD.
Referências: Abaixo são apresentadas as referências quanto às famílias estudadas por Sousa PS (1992), pelo autor da presente tese, e a caracterização molecular da família GC1. Dados adicionais devem ser obtidos nesses originais.
Cassa E. Ataxia Cerebelar Autossômico Dominante (ACAD) no Brasil: Análise de 270 anos de história e genealogia, incluindo a caracterização molecular de uma grande família com doença de Machado-Joseph. Tese apresentada à Faculdade de Medicina de Ribeirão Preto para obtenção do título de doutor em Medicina. Ribeirão Preto, São Paulo 1996.
Link para download em: https //drive.google.com/file/d/18IfAzgZq3E4SyGT-g_D84qVlqHJZozcZ/view?usp=sharing
Igarashi S et al (1996), Takiyama Y, Cancel G et al (1996). Intergenerational instability of the CAG repeat of the gene for Machado-Joseph Disease (MJD1) is affected by the genotype of the normal chromosome: implications for the molecular mechanisms of the instability of the CAG repeat. Human Molecular Genetics 5 (7): 923-932.
Sousa PS. Ataxia cerebelar autossômica dominante. Estudo de 11 famílias brasileiras. Dissertação apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo, para obtenção do título de mestre em Neurologia. Ribeirão Preto, São Paulo 1992.
Stevanin G, Cassa E, Cancel G, Abbas N, Dürr A, Jardim E, Agid Y, Sousa PS & Brice A (1995a) Characterization of the unstable CAG repeat in the MJD1 gene in four Brazilian families of Portuguese descent with Machado-Joseph disease. Journal of Medical Genetics 32 (10): 827-830
Stevanin G, Cancel G, Didierjean, Durr A, Abbas A, Cassa E, Feingold J, Agid Y, Brice A (1995b). Linkage disequilibrium at the Machado-Joseph disease/Spinal cerebellar ataxia 3 locus: Evidence for a common founder effect in french and portuguese-brasilian families as well as a second ancestral Portuguese-Azorean mutation. American Journal of Human Genetics 57: 1247-1250
Stevanin G, Sousa PS, Cancel G, Durr, Duborg O, Nicholson GA, Weissenback J, Jardim E, Agid Y, Cassa E, Brice A (1994). The gene for Machado-Joseph disease maps to the same 3 cM interval as the spinal cerebellar ataxia 3 gene on chromosome 14q.Neurobiology of Disease 1: 79-82