Polineuropatias: Apresentação clínica
Resumo. Um algoritmo para o diagnóstico das polineuropatias, baseia-se em primeiro lugar em separá-las das neuropatias multifocais, em seguida caracterizar se o curso clínico da doença é agudo, subagudo, ou crônico, e em seguida estabelecer se a ENMG é do tipo axonal ou desmielinizante. As polineuropatias subagudas e crônicas representam maior contingente de polineuropatias, devendo portanto ser bem identificadas pela sua apresentação simétrica e distal, iniciando-se com sintomas sensitivos pelos membros inferiores, com ou sem sintomas motores e ENMG axonal ou desmielinizante uniforme; estas neuropatias são relacionadas a doenças hereditárias e adquiridas as quais devem ser investigadas. Também dentro do grupo agudo e subagudo (grupos G2, G3, G5 das causas) encontra-se a polineuropatia idiopática ou criptogenética (grupo G6), com quadro clínico similar. A segunda apresentação clínica importante são as neuropatias inflamatórias desmielinizante (NID), cujo representante mais típico é a síndrome de Guillain-Barré (NIDA). A SGB-NIDA caracterizam-se por um déficit motor progressivo e reversível, distal (polineuropatia) e proximal (radiculopatia), poucos sintomas sensitivos, elevação proteica no LCR e ENMG desmielinizante não uniforme.
Algorítmo diagnóstico das neuropatias difusas. No algoritmo para o diagnóstico das neuropatias difusas (tabela 1), elas são subdivididas em neuropatia multifocal e polineuropatia. Em nossa opinião o caminho mais curto e mais prático para o diagnóstico é feito em 4 etapas: 1) avaliar se a apresentação clínica (Barohn RJ et al., 2013) é de início agudo (dias a < 4 semanas), subagudo (4-8 semanas) ou crônico (> 8 semanas); 2) as neuropatias subagudas e crônicas representam maior contingente de polineuropatias (grupos G2, G3, G5 e G6), devendo portanto ser bem identificadas; 3) Também nas formas subagudas e crõnica está incluído a polineuropatia axonal idiopática (PAIC no grupo G6 das causas)) com quadro clínico similar, mas para a qual não foi identificado uma etiologia conforme proposto no capítulo de causas; 4) reconhecer o quadro clínico das neuropatias inflamatória desmielinizantes (NID) as quais apesar de terem várias formas clínicas manifestam-se na maioria com alteração distal e proximal, predominando um déficit motor progressivo, elevação proteica no LCR e ENMG desmielinizante não uniforme; 6) o padrão da ENMG, se axonal, desmielinizante uniforme (DU), desmielinizante não uniforme (DND), e, caso a ENMG seja normal, avaliar a possibilidade de polineuropatias de fibras finas. As alterações na ENMG são avaliadas em um capítulo à parte.
 |
Polineuropatias Agudas. O exemplo mais típico de neuropatia aguda é a síndrome de Guillain-Barré (SGB) ou neuropatia inflamatória desmielinizante aguda (NIDA). Aquí consta uma revisão sumária do quadro clínico a ser mais detalhado em um capítulo específico.
A síndrome de Guillain-Barré. A SGB-NIDA (Grupo G1 das causas) caracteriza-se principalmente por um déficit motor progressivo atingindo seu máximo em até 4 semanas, sendo simétrico, distal e proximal e associado a reflexos diminuídos ou ausentes. Aquí tratamos do quadro clínico geral da SGB-NIDA, pois, seu diagnóstico encontra-se discriminado em etapas num capítulo a parte. A ENMG mostra desmielinização não uniforme (tipo adquirido) e o LCR uma elevação proteica com poucas células (dissociação albuminocitológica); a SGB também apresenta uma forma axonal com clínica similar à desmielinizante.O termo SGB, é equivalente a polirradiculoneuropatia e NIDA, os quais são termos complexos e relativos a diferentes épocas de caracterização desta doença; polirradiculoneuropatia aquí significa que a SGB além de envolver os nervos periféricos como as demais neuropatias, também envolve as raízes destes nervos, levando a déficit de força distal (perna e pé) e proximal (coxa e ombro). NIDA, é um termo mais recente e relacionado à etiologia inflamatória e desmielinizante. No algoritmo (tabela 1) as principais polineuropatias a serem diferenciadas da SGB são a porfiria, a neuropatia da doença crítica (UTI) e as vasculites; todas estas neuropatias são do tipo axonal incluindo também a SGB forma mais rara do tipo axonal. Embora a SGB seja a mais comum das polineuropatias agudas é importante diferenciá-la da porfiria aguda e da polineuropatia das doenças críticas.
Porfiria aguda. É uma neuropatia mais rara dentro dos grupos etiológicos, mas é a principal entidade a ser diferenciada da NIDA. A neuropatia ocorre nas formas porfiria aguda intermitente e porfiria variegata. O quadro clássico e completo associa dor abdominal aguda, náuseas, vômitos, diarréia, agitação mental e delírio; a neuropatia desenvolve-se 2-3 dias após os sintomas iniciais, com progressão variável (dias a semanas). O déficit motor desta polineuropatias pode ser predomínio distal ou proximal, podendo também iniciar pelos membros superiores; estes achados fazem exceção ao predomínio distal visto nas polineuropatias tipo axonal. Os reflexos são diminuídos proporcionalmente ao déficit motor, sintomas sensitivos não são proeminentes e falência respiratória pode ocorrer nos quadros graves; o LCR (diferentemente da SGB), não apresenta elevação proteica importante, a ENMG mostra lesão axonal e o diagnóstico é confirmado pelos níveis elevados de porfobilinogênio e ácido aminolevulínico urinários.
A polineuropatia de doenças críticas. A PDC surge em ambiente de terapia intensiva, sepse, falência de múltiplos órgãos, ou seja quando impera a anarquia metabólica. Embora a avaliação clínica dos pacientes com PDC seja limitada pelo ambiente da UTI e pela anarquia metabólica que produz entorpecimento da consciência, é entretanto possível, constatar uma fraqueza generalizada, poupando nervos cranianos ou com uma paresia facial leve. Acredita-se que a etiologia desta polineuropatia dependa de vários fatores haja visto a situação crítica em que surge. Embora a PDC possa ter semelhança com a SGB é importante lembrar que os pacientes com SGB em geral vão para a UTI em virtude de complicações respiratórias e que a PDC se instala na UTI. Também pode acontecer que durante a internação na UTI, o déficit não seja notado, e após a alta hospitalar, o paciente procure um neurologista em virtude de adormecimento (perda de sensibilidade) e dificuldades motora distalmente nos membros inferiores. Diferentemente da SGB, na PDC o LCR não apresenta dissociação albuminocitológica, e a ENMG mostra lesões axonais com redução marcada na amplitude distal.
Polineuropatias Subagudas. Doenças metabólicas, doenças sistêmicas, efeito adverso de medicamentos e doenças hereditárias são as principais causas deste grupo de polineuropatia, no qual encontram-se o maior contingente de polineuropatias; destas polineuropatias, o diabetes mellitus é a causa mais frequente. Estas neuropatias são analisadas nos grupos G2, G3, G5 e G6 do capítulo causas e diagnóstico, e, a polineuropatia diabética em um capítulo específico. Estas polineuropatias apresentam-se inicialmente com sintomas sensitivos nos pés tipo adormecimento distal, formigamentos, pinicação, agulhamentos, etc. Sintomas motores podem não ocorrer, assim como podem se manifestar mais tardiamente. Estas polineuropatias deste grupo são predominantemente do tipo axonal, com predomínio sensitivo e mais raramente sensitivo e motor. Uma menor proporção de casos a polineuropatia é desmielinizante e mais raramente pode comprometer apenas as fibras finas, e neste caso a ENMG é normal. Fazem exceção a regra descrita as neuropatias inflamatórias desmielinizante subaguda, cujo quadro clínico é dominado por uma paralisia progressiva e a ENMG mostra lesão desmielinizante.
Polineuropatia crônica. A separação entre polineuropatia subaguda e crônica é em parte artificial uma vez que neuropatias crônicas são aquelas que apresentam manifestações que foram sendo acumuladas ao longo de anos e décadas. O protótipo destas polineuropatias são as as neuropatias hereditárias sensitivo e motoras (NHSM) ou doença de Charcot Marie Tooth (NHSM-CMT). A diferença fundamental entre estes tipos é que nas polineuropatias subagudas os sintomas sensitivos e motores são uma novidade enquanto nas crônicas eles vão se superpondo lentamente ao longo de anos ou mesmo décadas e em muitos casos não são percebidos e/ou são assintomáticos.
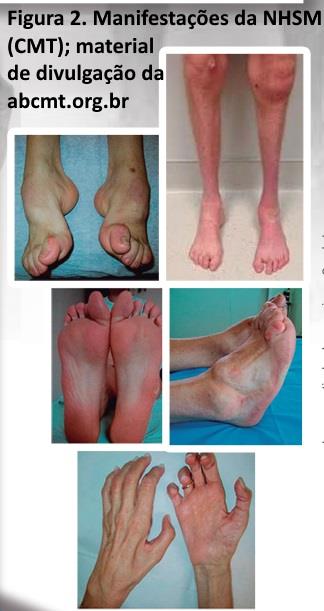
As NSMH-(CMT). Apresentam uma evolução lentamente progressiva, onde os sinais clínicos florescem durante anos ou mesmo décadas, resultando em manifestações tróficas características (estigmas), tais como pé cavo, dedos em martelo, calos plantares e dedos da mão em garra (figura 2). Quando o quadro da NSMH-(CMT) encontra-se avançado, os membros inferiores apresentam-se atrofiados distalmente, mostrando um aspecto de uma garrafa invertida; um déficit motor distal dos membros inferiores e um pé caído também podem ser encontrados. Para confirmar este diagnóstico é importante obter tanto dos pacientes como de familiares (com permissão), uma história, exame clínico e dados adicionais (fotografias por exemplo). Importante executar este procedimento já na anamnese, pois, uma vez o diagnóstico de NSMH (CMT) seja perdido no início, uma cascata de exames podem ser agendados de forma onerosa.
Os achados na família variam de leves a severos na dependência da duração da doença em cada indivíduo. Embora as neuropatias tipo
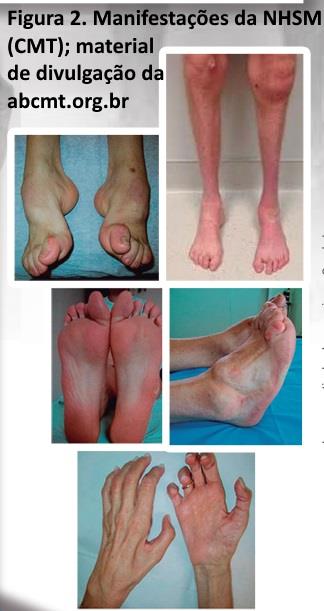 |
NHSM (CMT) sejam na prática diária um exemplo de uma neuropatia crônica, nada porém impede que uma neuropatia adquirida (diabetes ou alcoolismo por exemplo) apresente manifestações similares (embora menos severas), a longo prazo. Detalhes genéticos das CMT (NHSM) podem ser obtidos na na associação Brasileira dos portadores de CMT (ABCMT) e na neuromuscular home page com links ao final do capítulo. As NHSM (CMT) podem ser do tipo axonal ou desmielinizante uniforme.
Polineuropatia idiopática crônica (PAIC). Dentro das polineuropatias crônicas é importante o reconhecimento da PAIC, a qual corresponde a aproximadamente 20% do total das polineuropatias. O termo idiopática refere-se a não identificação de uma causa após a investigação proposta no capítulo de causas e diagnóstico. A PAIC têm muitos aspectos similares à polineuropatia sensitiva distal do diabetes e das polineuropatias subagudas e crônicas. A PAIC predomina em paciente com 40 a 70 anos, é insidiosa, distal simétrica, manifestações sensitivas predominam sobre sensitivo-motora, inicia pelos membros inferiores e atinge os membros superiores em aproximadamente 5 anos. Os principais sintomas são aparecimento, parestesias e dor; cerca de 60% dos casos de PAIC terão dor neuropática e 10% terão apenas dor. No exame clínico encontramos redução das sensibilidade vibratória (80-100%), proprioceptiva (20-30%), dolorosa (70-85%) e tátil (50-94%). A força muscular em geral é normal, podendo entretanto haver déficit motor e atrofia distal nos MMII, associada a reflexos ausentes no tornozelo e diminuídos nos demais músculos. Muitos pacientes com PAIC apresentam neuropatia de fibras finas, com parestesias, adormecimento, redução na sensibilidade tátil e dolorosa, mas preservando a função das fibras grossas, cursando então com uma ENMG normal (figura 1); neste caso pode ser indicado uma biópsia de pele para análise das fibras finas.
Referências
Associação Brasileira dos Portadors de Charcot-Marie-Tooth (ABCMT): http://abcmt.org.br/
Barohn RJ, Amato AA. Pattern-recognition approach to neuropathy and neuronopathy. Neurol Clin. 2013 May;31(2):343-61.
Herskovitz S, et al (2010). Evaluation and management of patients with peripheral neuropathy. In: Herskovitz S. et al (2010) Peripheral neuropathy in clinical practice, Oxford University Press. pg 24-39
Herskovitz S, et al (2010). Evaluation and management of patients with peripheral neuropathy. In: Herskovitz S. et al (2010) Peripheral neuropathy in clinical practice, Oxford University Press. pg 24-39
Lubec D, Müllbacher W, Finsterer J, Mamoli B. Diagnostic work-up in peripheral neuropathy: an analysis of 171 cases. Postgrad Med J. 1999 Dec;75(890):723-7.
Pasnoor M, Nascimento OJ, Trivedi J et al. North America and South America (NA-SA) neuropathy project. Int J Neurosci. 2013 Aug;1
Willison HJ, Winer JB. Clinical evaluation and investigation of neuropathy. J Neurol Neurosurg Psychiatry. 2003 Jun;74 Suppl 2:ii3-ii8.