Mestrado: História Natural da Síndrome de Guillain-Barré em Crianças
 |
Resumo. A síndrome de Gillain-Barré representa uma doença do sistema nervoso periférico secundária a inflamação com desmielinização. A SGB típica inicia-se com uma paralisia dos membros inferiores que progride podendo comprometer todo o corpo incluindo parada respiratória. Neste capítulo apresentamos a história natural da síndrome de Guillain-Barré em 24 crianças (10M, 14 F) os quais na época de instalação da síndrome tinham a idade média de 6 anos. Durante as manifestações agudas da síndrome a maioria (20 pacientes) apresentou um déficit motor progressivo e severo, ficando incapacitados para deambular, incluindo 2 casos que apresentaram falência respiratória, sendo mantidos em ventilação mecânica; não houve óbitos durante a síndrome. Foi determinado que recuperação na SGB encerra-se após 2 anos do início do quadro e nesta época as sequelas presentes serão definitivas; todos os pacientes apresentaram boa recuperação, retornando às suas atividades diárias após 1 ano do início do quadro e após 2 anos, apenas 4 pacientes apresentavam sequelas leves nos membros inferiores. Adicionalmente após um seguimento ambulatorial de 4.3 anos em média, não foi observado nestes pacientes, comorbidades importantes ou patologias adicionais (novas). Comparada a literatura relativa a SGB em crianças (SGBC) e adultos (SGBA), o prognóstico da nossa casuística, apresenta duas conclusões: 1) Uma baixa prevalência de sequelas graves após 2 anos, fato que concorda com a literatura da SGBC, mas, a diferencia da SGBA onde a prevalência de sequelas graves é maior; 2) Diferentemente da literatura nossa casuística não registrou mortalidade, fato presente em 6-7% da literatura (SGBA + SGBC). O link para download da tese encontra-se nas referências ao final do capitulo.
Desenho do estudo. Com os objetivos de descrever a história natural e o prognóstico da síndrome de Guillain-Barré em crianças (SGBC), foram estudados 24 pacientes (14M e 10F), os quais foram seguidos no serviço de neurologia da Faculdade de Medicina de Ribeirão Preto (USP), no período de 1979 a 1990. O estudo inclui a fase aguda da doença (idade média de 6.08 anos), o seguimento ambulatorial (4.3 anos em média), até o exame de reavaliação (idade média de 10.7 anos). Os pacientes foram incluídos em acordo com os critérios de Asbury (1978). O déficit motor foi estadiado (E) por todo o período de estudo segundo a escala de Hughes (1978), com modificações (tabela 1).
A avaliação complementar constou, na fase aguda de exame de líquido cefalorraquidiano (LCR) para todos os pacientes e eletroneurografia para 6 pacientes; no exame de reavaliação foi feito eletroneuromiografia em 14 pacientes.
 |
Manifestações agudas. Antecedendo o início do déficit motor por 11,8 dias em média, ocorreu um infecção prévia em 14 casos (58.33%) localizando-se predominantemente em vias aéreas superiores e trato gastrointestinal. Os primeiros sintomas da doença foram sensitivos em 14 casos (58.33%) predominantemente dolorosos, localizando-se nos membros inferiores ou ocorrendo de forma difusa, em ambos os casos piorando com esforço.
O déficit motor iniciou-se nos membros inferiores em 20 casos (83,33%) sendo ascendente em 18 (75%), em 3 casos o início foi pelos membros superiores. O déficit máximo foi atingido em menos de quatro semanas para todos os pacientes, sendo simétrico do tipo flácido e com arreflexia universal ou arreflexia distal com hiporreflexia proximal em 20 casos (83.33%). O déficit permaneceu estável por um tempo de 12 dias em média e segundo a escala de Hughes os pacientes tiveram um déficit médio de 3.88 estádios sendo distribuídos como: 18 casos (75%) em estádio 4, dois casos em estádio 5, e 4 casos em estádio 3 ou menos. Topograficamente o déficit foi de predomínio crural e braquial em 20 casos (83.33%) os demais tendo acometimento de predomínio crural.
Os nervos cranianos foram comprometidos em 18 casos (86.88%), 12 casos (50%) dos IX e X nervos, 6 casos (25%) do VII nervo e o VI nervo unilateral em um paciente. A sensibilidade pode ser determinada em 8 pacientes, dos quais 7 tinham comprometimento distal das sensibilidades tátil, dolorosa, vibratória e de posição segmentar sendo do tipo bota ou bota e luva.
As principais intercorrências foram respiratórias e disautonômicas. Ocorreu pneumonia em 4 casos, em dois destes casos que estavam em ventilação mecânica, a pneumonia foi acompanhada transitoriamente por atelectasia lobar. Disautonomias ocorreram em 20 casos (83.33%) principalmente como taquicardia em 12 casos (50%), disfunção em esfíncter vesical e constipação intestinal em 6 casos (25%) e hipertensão arterial e incontinência fecal em 3 casos.
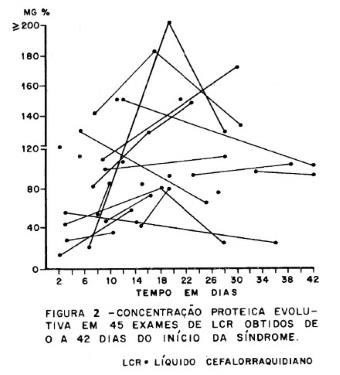 |
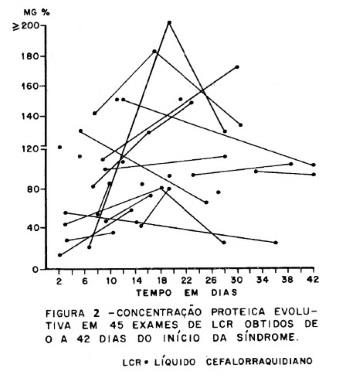
Exames complementares. O LCR e a eletroneurografia foram de grande valor diagnóstico; no LCR (figura 2) observou-se dissociação proteinocitológica em 23 casos (95.83%) sendo que a proteinorraquia mostrou um padrão evolutivo com níveis máximos do décimo ao vigésimo dia do início da síndrome, sendo a maior elevação de 310 mg. A eletroneuromiografia foi consistente com diagnóstico de neuropatia desmielinizante em 5 dos 6 casos em que foi executada. Considerados conjuntamente, o diagnóstico clínico foi feito em 20 casos (83.33%), clínico e pelo LCR em 24 casos (100%), clínico e eletroneurográfico em 22 casos (91.86%) e pelos LCR e eletroneurográfico em 23 casos (95 83%).
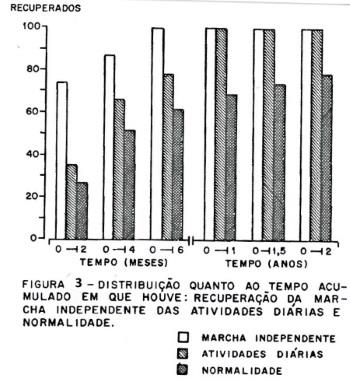
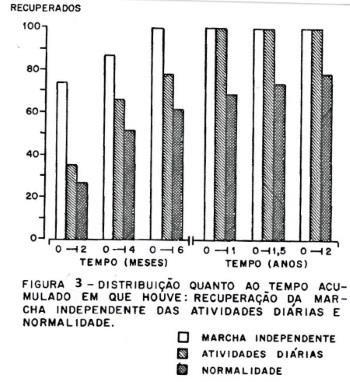
A recuperação. Na recuperação (figura 3), a marcha independente ocorreu em todos os pacientes (100%) até os 6 meses, o retorno às atividades diárias ocorreu em 23 casos (100%) até 1 ano e o retorno à normalidade que ocorreu em 17 casos (73,91%) até 1 ano, e em 18 casos (78.26%) aos 2 anos. Considerando a normalização aos 6 meses tivemos dois grupos de recuperação, um grupo com recuperação rápida com 15 pacientes normais aos 6 meses e um grupo com recuperação lenta com 8 pacientes que não atingiram a normalização aos 6 meses. A diferença significativa entre os dois grupos foi dada por um período de "plateau" mais prolongado no grupo de recuperação lenta. O ponto final da recuperação se deu aos 2 anos; cinco pacientes não atingiram a normalização num tempo de 3 a 9 anos
 |
Sequelas. O exame de reavaliação foi feito em 20 dos 24 pacientes, evidenciando que 15 estavam normais e 5 tinham seqüelas mínimas caracterizada por um déficit motor (paresia) distal simétrica leve nos membros inferiores (em 5 casos) e nos membros superiores (3 casos). Outros achados menos importantes foram: pé cavo, cifose dorsal, hiperextensão dos joelhos, hipotonia global, câimbras frequentes nos membros inferiores, hiperidrose difusa e hipopalestesia distal nos membros inferiores. O exame eletroneuromiográfico feito em 14 pacientes, mostrou em 12 destes, sequelas do tipo axonal predominando nos membros superiores, com redução de velocidade e amplitude dos dos potenciais sensitivos.
Revisão da literatura. Comparada a literatura relativa a SGB em crianças (SGBC) e adultos (SGBA) o prognóstico da nossa casuística, apresenta 2 conclusões: 1) uma baixa prevalência de sequelas graves após 2 anos, fato que concorda com a literatura da SGBC, e a diferencia da SGBA onde a prevalência de sequelas graves é maior; 2) Diferentemente da literatura nossa casuística não registrou mortalidade, condição presente em 6-7% da literatura (SGBA + SGBC).
Comentário: Em função de inúmeros pedidos, o resumo aqui apresentado foi modificado pelo autor, relativamente a tese original. Os diversos pedidos solicitaram uma apresentação mais popular e menos científica
Referências
Cassa E. História Natural da Síndrome de Guillain-Barré em Crianças. Tese apresentada ao departamento de neurologia, da Faculdade de Medicina de Ribeirão Preto (USP), para obtenção do título de mestre em medicina. Ribeirão Preto São Paulo 1991.
Link para download: https://drive.google.com/file/d/14KcKCHeduQkp_ofFSp8cn2Ax9PcuhUfn/view?usp=sharing